Sub-Total: $0.00 USD
Have questions about this assay kit?
Human PPARγ Reporter Assay Kit
Product Description and Product Data
This is an all-inclusive cell-based luciferase reporter assay kit targeting the Human Peroxisome Proliferator-Activated Receptor Gamma. INDIGO’s Human PPAR gamma reporter assay utilizes proprietary mammalian cells that have been engineered to provide constitutive expression of the Human PPAR gamma. In addition to PPAR gamma Reporter Cells, this kit provides two optimized media for use during cell culture and in diluting the user’s test samples, a reference agonist, Luciferase Detection Reagent, and a cell culture-ready assay plate. The principal application of this assay is in the screening of test samples to quantify any functional activity, either agonist or antagonist, that they may exert against human PPAR gamma.This kit provides researchers with clear, reproducible results, exceptional cell viability post-thaw, and consistent results lot to lot. Kits must be stored at -80C. Do not store in liquid nitrogen. Note: reporter cells cannot be refrozen or maintained in extended culture.
Features
Ready to Use Upon Receipt
- Includes All Needed Components
- Contains Transfected Reporter Cells
- Eliminates Cell Licensing Fees
- Clear, Reproducible Results
- Consistent Results Lot to Lot
Product Specifications
| Target Type | Nuclear Hormone Receptor | ||
| Species | Human | ||
| Receptor Form | Hybrid | ||
| Assay Mode | Agonist, Antagonist | ||
| Kit Components |
| ||
| Shelf Life | 6 months | ||
| Orthologs Available | Yes | ||
| Shipping Requirements | Dry Ice | ||
| Storage temperature | -80C |
Data


Target Background
Peroxisome Proliferator-Activated Receptor Gamma (PPARγ), also known as the glitazone receptor, or NR1C3 is a type II nuclear receptor that in humans is encoded by the PPARγ gene. PPARs form heterodimers with Retinoid X Receptors (RXRs) and these heterodimers regulate transcription of various genes. PPARγ regulates adipocyte differentiation, fatty acid storage and glucose metabolism. The PPARγ knockout mice fail to generate adipose tissue when fed a high fat diet. Many insulin sensitizing drugs used in the treatment of diabetes target PPARγ as a means to lower serum glucose without increasing pancreatic insulin secretion. Additionally, PPARγ has been implicated in the pathology of numerous diseases including obesity, diabetes, atherosclerosis and cancer. Alternatively spliced transcript variants that encode different isoforms have been described.
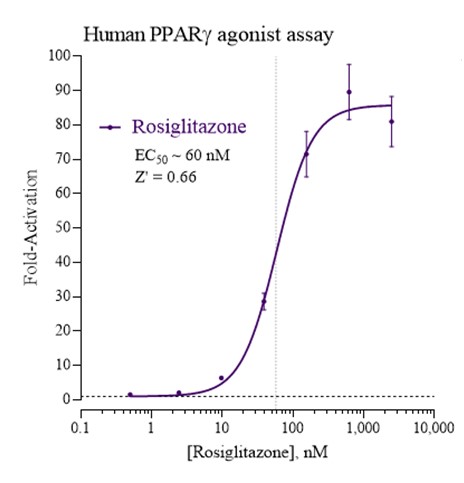
INDIGO’s PPARγ Reporter Assay System utilize proprietary mammalian cells engineered to express human PPARG, commonly referred to as PPARγ.
The principle application of this assay product is in the screening of test samples to quantify functional activities, either agonist or antagonist, that they may exert against the human peroxisome proliferator-activated receptor gamma.
Citations
Also available as a service